家族性ALS原因遺伝子産物である変異型SOD1によって引き起こされる神経軸索輸送障害
疾病研究第五部
SOD1 (スーパーオキシドジスムターゼ1)は、家族性筋萎縮性側索硬化症(Amyotrophic lateral sclerosis, ALS;運動神経細胞が特異的に変性する致死性の神経難病)の原因遺伝子の一つです。現在日本には約1万人のALS罹患者がおられますが、有効な治療法はなく、新たな治療法開発が強く望まれています。
我々の研究室では、患者から同定されたヒト変異型SOD1タンパクを発現する疾患モデル動物(マウス)を用いて、変異型SOD1タンパクが脊髄運動神経細胞に障害を引き起こすメカニズムを調べていました。これまでの研究から、変異型SOD1は異常な立体構造をもち、凝集化してしまうことで細胞毒性を発揮すると考えられます。我々は今回、構造異常を伴うSOD1タンパクが神経軸索輸送関連分子の一つであるキネシン2の機能を特異的に阻害するために、脊髄運動神経の伝達物質合成に必須の酵素(コリンアセチル基転移酵素)の神経の末端への輸送ができなくなることを明らかにしました。
手足の筋肉を直接支配する運動神経は脊髄に細胞体があり、そこから筋肉まで長い突起(軸索)を伸ばし、突起の先端で筋肉組織と連絡しています。大脳などからの指令によって、運動神経細胞は末端からアセチルコリンを放出し、それに伴って筋肉の収縮が起こることによって随意運動が可能になります。その後、アセチルコリンは速やかに再吸収・分解されることで運動は終了し、神経末端において「コリンアセチル基転移酵素」と呼ばれる酵素の働きによってアセチルコリンへと再合成されます。従って神経伝達にはコリンアセチル基転移酵素が神経終末に十分量存在することが必要です。この酵素は細胞体で作られるため、神経終末まで輸送される必要があります。神経細胞は多種類の分子によってさまざまな物質を神経終末へと輸送していますが、どの分子が何を運ぶかは決まっており、コリンアセチル基転移酵素の輸送は「キネシン2」によって行われていることが報告されていました。
我々は疾患モデルマウスにおいてはALSの運動症状が発症するかなり前から構造が異常化したSOD1タンパクが運動神経の軸索内に存在し、かつ末端に向かって移動していることを見つけ、構造が異常化したSOD1タンパクによる軸索輸送障害の可能性を考えました。そこで、発症期のマウスの脊髄白質(神経突起の存在する部位)の抽出液から構造が異常化したSOD1を分離し、17種の軸索輸送関連分子について構造が異常化したSOD1との結合の有無を調べたところ、キネシン2の構成成分であるKAP3が、構造の異常化したSOD1と特異的に結合していることがわかりました。
KAP3はキネシン2の構成成分の中で、キネシン2によって輸送される積荷(担体)を認識して直接結合する役割をもち、言わば何を輸送するかを決定する役目を担っています。上述のように、キネシン2が輸送する担体としてコリンアセチル基転移酵素があります。我々は、モデルマウスの脊髄運動ニューロン軸索中では、SOD1の構造異常化とともにコリンアセチル基転移酵素の輸送量が低下することを見出しました。また、運動神経様に分化させた培養細胞を用いた実験では、変異型SOD1の構造異常化を誘導すると、電気的興奮により神経末端から放出されるアセチルコリンの量が減少することを認めました(図1)。このことは、構造異常化したSOD1とKAP3の結合によってキネシン2の機能が阻害(コリンアセチル基転移酵素などの輸送が阻害)されたことが原因と考えられます。このような異常なSOD1とKAP3の特異的な結合は、モデルマウスだけでなく、SOD1変異を有する家族性ALS患者様の脊髄運動神経細胞においても高頻度に起きていることがわかりました(図2)。
家族性・孤発性を問わず、ALSでは発症期には既に神経伝達が低下していることが報告されています。今回の我々の発見は、SOD1変異による家族性ALSにおいては、構造異常化したSOD1タンパクによるキネシン2活性の阻害が(コリンアセチル基転移酵素の輸送障害などを介して)神経伝達の低下をもたらすことを強く示唆するものです(図3)。
ALSの大部分を占める孤発性(遺伝的な変化を伴わない)ALS患者様の大脳運動野ではキネシン2の発現量が特異的に減少している、という報告もなされていることから、キネシン2の阻害は家族性・孤発性に共通したALS発症への経路である可能性もあり、今後更に検討を行っていく予定です。
Tateno M., Shinsuke Kato S., Sakurai T., Nukina N., Takahashi R., and Araki T. Mutant SOD1 impairs axonal transport of choline acetyltransferase and acetylcholine release by sequestering KAP3.
Human Molecular Genetics, 18: 942-955 (2009)
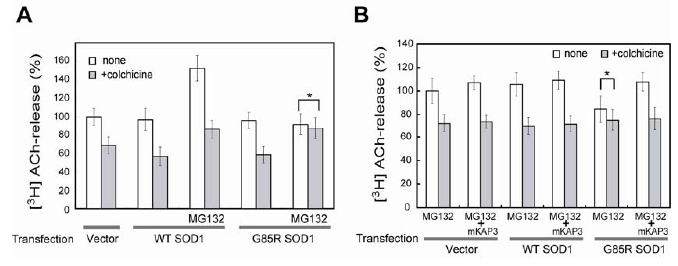
図1 ミスフォールド化SOD1による軸索依存性アセチルコリン放出の減少 [図を拡大する]
PNG3細胞にvector、野生型SOD1(WT)、変異型SOD1(G85R)発現ベクターを導入し、コリン作動性ニューロン様へ分化誘導後3日目に培地に[3H]コリン(アセチルコリンの前駆体)を添加し、50 mM KClによって誘発される[3H]アセチルコリン放出量を測定した。白色バー、灰色バーはそれぞれ、微小管重合阻害剤であるコルヒチン非添加群(総放出量)、添加群(軸索に依存しない放出量)を示しており、両者の差が軸索に依存したアセチルコリン放出量である。変異型SOD1タンパクのミスフォールド化はMG132(プロテアソーム阻害剤)処理によって誘導した。なお、白色バー・灰色バーの比較において有意差のない場合(p>0.05)だけ*で示す。A)変異型SOD1導入後、ミスフォールド化を誘導した場合においてのみ、軸索依存性のアセチルコリン放出分が消失する(*: p>0.05)。B)Aで消失した軸索依存性アセチルコリン放出が、KAP3の過剰発現によって復帰。
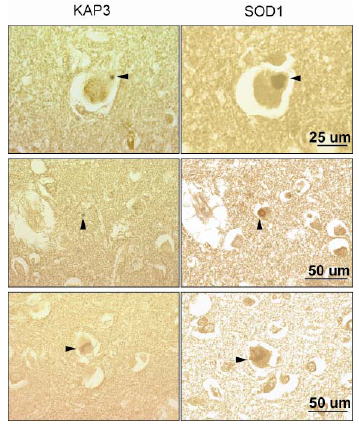
図2 ヒト家族性ALS脊髄運動ニューロン内におけるSOD1凝集体とKAP3の共局在
SOD1変異を有する患者3例の脊髄連続切片を抗KAP3抗体、抗SOD1抗体で染色。ALS患者組織に特徴的に認められる封入体(Lewy-body-like hyaline inclusions, LBHIs)内に存在するKAP3, SOD1凝集像を矢印で示した。LBHIs内のほとんどで両者は共局在していた。
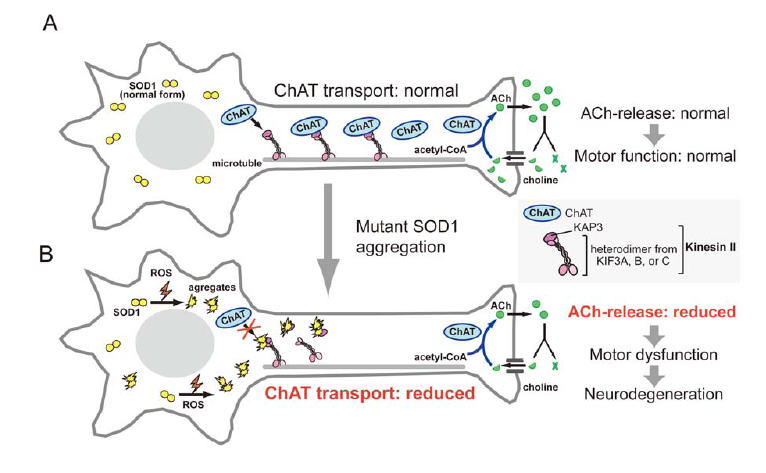
図3 変異型SOD1による軸索輸送障害 [図を拡大する]
A)健全な運動ニューロンでは、細胞体で合成されたコリンアセチル基転移酵素(ChAT)はKAP3(キネシン2の担体結合サブユニット)との結合を介してキネシン2により軸索内を神経終末へと輸送される。神経終末では刺激に伴いアセチルコリンの放出・分解・再吸収・再合成が盛んに行われており、コリンアセチル基転移酵素はこのサイクル反応の律速酵素である。B)酸化ストレスの増大などにより変異型SOD1タンパクのミスフォールド化が促進されると、ミスフォールド化SOD1がKAP3と特異的に結合するためにキネシン2は本来の輸送担体と結合できなくなる。その結果、コリンアセチル基転移酵素の輸送量が減り、神経終末におけるアセチルコリンの供給低下、ひいては神経伝達の低下をもたらす。