Our Research
Our research aims to develop therapeutic aproaches for neurlological disorders such as neurodegenerative diseases through understanding molecular mechanism of axonal degeneration and regeneration, and de-myelination and re-myelinataion.
Subcellular signaling that leads to degeneration of neurons and nerves is shared in part with signaling that mediates neuronal development and differentiation. This type of signaling may be constantly affected by physiological activities involving modification of energy metabolism and cellular redox status. We aim to find protect neurons and nerves from degeneration and to maintain functionality of the nervous system throughout longevity in a mild way via modification of cellular signaling elicited by physiological behaviors.
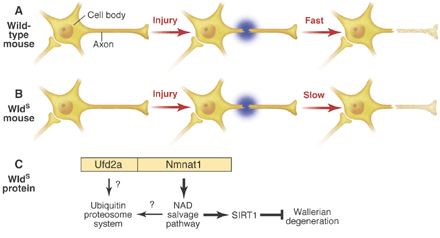
Aim 1: To clarify the mechanism of axonal degeneration
Axonal degeneration often precedes the death of neuronal cell bodies in neurodegenerative disease. The prevention of axonal degeneration may therefore represent an important point of intervention in combating these diseases. The phenotype of wlds mutant mice, which have a delay in Wallerian degeneration, led to the discovery that axons degenerate through an active self-destructive program. We are trying to dissect the mechanism of axonal degeneration and to modify the degeneration process via pharmacological as well as molecular biological methods.
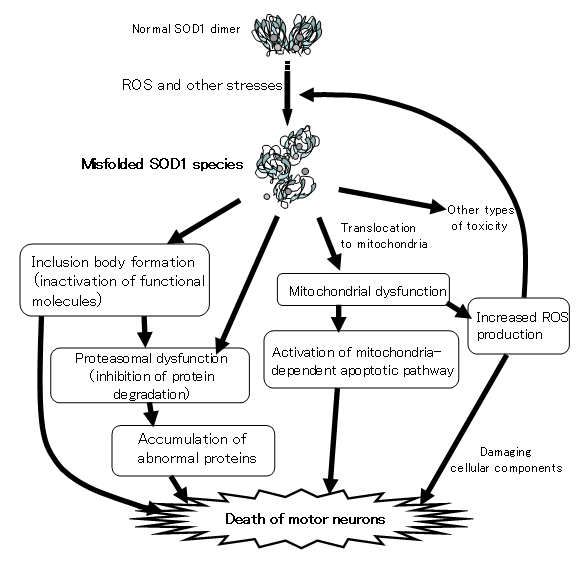
Aim 2: To identify initial process of degeneration of motor neurons in familial amyotrophic lateral sclerosis (FALS).
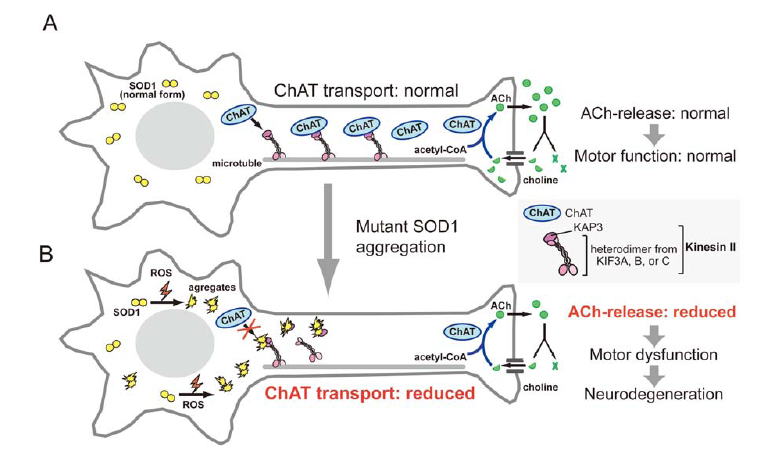
Mutations in the superoxide dismutase 1 (sod1) gene cause FALS, likely due to the toxic properties of misfolded mutant SOD1 protein. Misfolded SOD1 species specifically associates with Kinesin-associated protein 3 (KAP3) in the spinal cord of G93A SOD1-Tg mouse, a FALS model, from the pre-onset stage. KAP3 is a Kinesin-2 subunit responsible for binding to cargos including choline acetyltransferase (ChAT), whose transport was selectively reduced in G93A SOD1-Tg motor axons prior to the disease onset. We previously reported that KAP3 sequestration by misfolded SOD1 species and resultant inhibition of ChAT transport play a role in the pathogenesis of ALS. We are trying to show how important this mechianism is for initial motor dysfunction observed in the FALS animal model.
Mutant SOD1 toxicity: abnormal protein accumulation hypothesis [Image zoom]
A schematic diagram for a mutant SOD1 toxicity by impairment of axonal transport [Image Zoom]
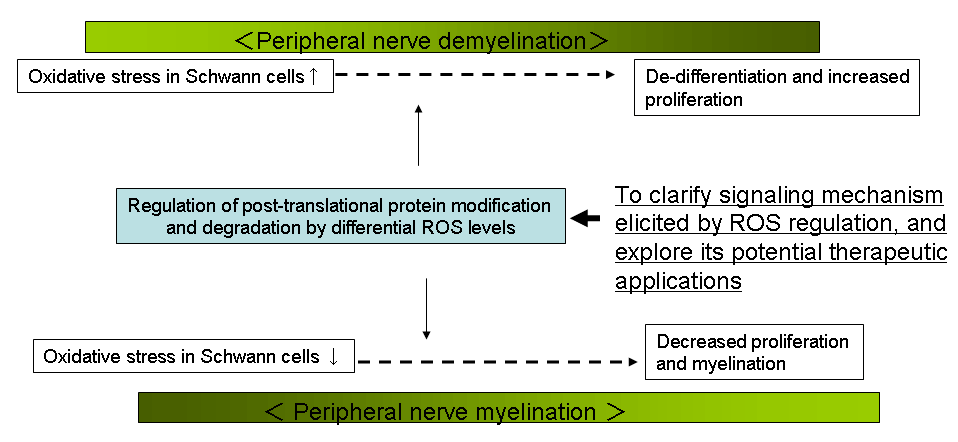
Aim 3: To understand mechanism of the peripheral nerve myelination and degeneration.
Defects in peripheral nerve regeneration and myelination due to either traumatic injury, exposure to toxins, or from acquired (e.g., diabetes) or hereditary (e.g., CMT) disease cause significant morbidity.
Using various algorithms to analyze expression profiles of injured sciatic nerve, we have identified a large number of transcripts in Schwann cells surrounding injured axons that are likely to be involved in myelination and nerve regeneration. Among genes induced in Schwann cells after nerve injury, we have identified a novel ubiquitin ligase, ZNRF1. ZNRF family of ubiquitin ligases are constitutively expressed in neurons. We are using molecular and cell biological techniques to understand the role of ZNRF family as well as significance of ubiquitination in presynaptic exocytosis as well as maintenance of axonal integrity.