

難病「ペリツェウス・メルツバッハ病」の原因に新説
― “細胞内カルシウム不足”が細胞内物流を停止、治療の標的に ―
国立精神・神経医療研究センター(NCNP)メディカルゲノムセンター 井上 健 室長らの研究グループは、遺伝性白質疾患であるペリツェウス・メルツバッハ病(PMD)1)の新たな発症メカニズムを明らかにしました。PMDは、ミエリン形成の異常により重篤な神経症状を呈する小児の希少疾患であり、これまで有効な治療法は確立されていません。従来は、変異PLP1タンパク質による「小胞体ストレス」が主因と考えられてきました。本研究では、細胞内の小胞体カルシウム濃度の低下がタンパク質輸送の障害を引き起こすことを培養細胞および疾患動物モデルを用いて明らかにし、さらにこの異常が薬剤により回復する可能性があることを示しました。
本成果は、神経科学系雑誌「Neurobiology of Disease」(4月号)に2026年2月24日に掲載されました。PMDの病態理解を大きく進展させるとともに、新たな治療戦略の開発につながることが期待されます。
研究成果のポイント
• 難病PMDの新たな発症メカニズムを解明
• 細胞内小胞体カルシウム低下 → タンパク質輸送停止を発見
• 従来の「小胞体ストレス中心」から「機能障害」への転換
• 薬剤で回復可能性を確認
• 新規治療標的として期待
• 細胞内小胞体カルシウム低下 → タンパク質輸送停止を発見
• 従来の「小胞体ストレス中心」から「機能障害」への転換
• 薬剤で回復可能性を確認
• 新規治療標的として期待
概要図
研究の背景
ペリツェウス・メルツバッハ病(PMD)は、PLP1遺伝子の変異により発症する中枢神経系の遺伝性疾患で、ミエリン形成不全(白質形成不全)を特徴とします。厚生労働省により難病(先天性大脳白質形成不全症)として指定されている疾患です。これまで、変異PLP1タンパク質が小胞体内に蓄積し、「小胞体ストレス」を引き起こすことが病態の中心と考えられてきました。
しかし、近年の研究では、小胞体ストレスを軽減しても病態が十分に改善しないことが示されており、別の重要な病態機構の存在が示唆されていました。
研究の内容
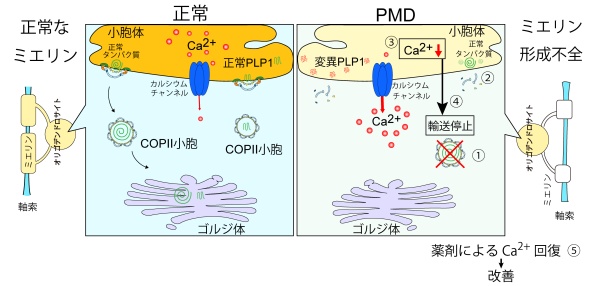
研究グループは、細胞モデルを用いて変異PLP1が細胞機能に与える影響を詳細に解析しました。その結果、下記を明らかにしました。• 細胞内(小胞体内)のカルシウム濃度が低下する
• COPII小胞(タンパク質輸送に必須)の形成が障害される
• 小胞体からゴルジ体への輸送が停止する
これは、細胞内でタンパク質が適切に運ばれない「細胞内物流の停止」ともいえる状態です。
本研究では、変異PLP1が細胞機能に与える影響を、複数の実験を用いて段階的に解析しました。下記にその詳細を説明します(カッコ内は前出の概要図の中に示された番号)。
タンパク質輸送の遅延(①)
まず、細胞内でのタンパク質輸送を解析したところ、変異PLP1を発現した細胞では、小胞体(ER)からゴルジ体への輸送が著しく遅延していることが分かりました。
具体的には、輸送マーカーであるVSV-Gタンパク質を用いた解析により、正常細胞では時間とともにゴルジ体へ移行するのに対し、変異PLP1発現細胞では輸送効率が大幅に低下していました。 これは、細胞内の「物流機能」が障害されていることを示します。
COPII小胞形成の異常(②)
次に、輸送の初期段階であるCOPII小胞形成を解析しました。免疫染色により、通常は小胞体出口部位(ERES)に集積するCOPII構成タンパク質(Sec31A、Sec24B)が、変異PLP1発現細胞では集積せず、散在していることが確認されました。 さらに細胞分画解析では、これらのタンパク質が本来存在すべき小胞体膜ではなく細胞質側に偏在していることが示されました。つまり「COPII小胞は作る材料はあるが、組み立てられない状態」であることが明らかになりました。
小胞体カルシウムの枯渇(③)
次に、COPII形成異常の原因としてカルシウムに着目しました。小胞体内カルシウム濃度を可視化したところ、変異PLP1発現細胞ではカルシウム濃度が正常の約35%まで低下していました。さらに、この現象は培養細胞だけでなく、マウス脳内のオリゴデンドロサイトにおいても確認されました。これは、PMDにおいて「カルシウム恒常性の破綻」が実際に起きていることを示しています。
カルシウム依存的な分子相互作用の破綻(④)
COPII小胞形成には、カルシウム依存的な分子複合体(ALG-2/Sec31A/AnxA11)が必要です。本研究では、これらの分子間相互作用が変異PLP1発現細胞では著しく低下していることを示しました。すなわち、カルシウム低下によりCOPII小胞形成の“足場”そのものが崩壊していることが分かりました。
カルシウム回復による機能改善(⑤)
さらに、薬剤により小胞体カルシウムを回復させると、することが確認されました。
• Sec31Aの局在(ERへの再集積)が回復
• COPII小胞形成が改善
• タンパク質輸送も回復
これは本研究の非常に重要な点で、病態が可逆的である可能性を示しています。
病態の重症度との相関
さらに、異なるPLP1変異を比較したところ、下記が、臨床症状の重症度と相関することが示されました。
• カルシウム低下の程度
• COPII形成障害の程度
すなわち本機構は、単なる現象ではなく病気の本質的なドライバーである可能性が高いと考えられます。
研究成果の意義
本研究は、PMDの病態の理解を従来の細胞死(小胞体ストレス中心)から細胞機能障害(輸送不全)へと大きく転換するものです。特に重要なのは、カルシウム異常が細胞機能障害の上流にある“制御可能な要因”である点です。さらに、カルシウムを回復させる薬剤により輸送障害が改善し、病態が可逆的である可能性を示唆したことから、治療標的としての実現可能性が高い発見といえます。
また、本メカニズムは他の遺伝性神経疾患にも共通する可能性があり、広範な応用が期待されます。
今後の展望
今後は、なぜ変異PLP1が小胞体のカルシウムを放出させるのか、その分子機序を解明するとともに、カルシウム制御を標的とした特異的な治療薬の開発を進め、臨床応用を目指します。論文情報
• 掲載誌:Neurobiology of Disease• タイトル:Mutant PLP1 impairs COPII vesicle formation via ER calcium depletion in Pelizaeus-Merzbacher disease
• DOI:10.1016/j.nbd.2026.107329



{kind=link}